

What are thalassemias?
Thalassemias (thal-a-SE-me-ahs) are inherited blood disorders. "Inherited" means they're passed on from parents to children through genes.
Thalassemias cause the body to make fewer healthy red blood cells and less hemoglobin (HEE-muh-glow-bin) than normal. Hemoglobin is an iron-rich protein in red blood cells. It carries oxygen to all parts of the body. It also carries carbon dioxide (a waste gas) from the body to the lungs, where it's exhaled.
People who have thalassemias can have mild or severe anemia (uh-NEE-me-uh). This condition is caused by a lower than normal number of red blood cells or not enough hemoglobin in the red blood cells.
What Causes Thalassemias?
Your body makes three types of blood cells: red blood cells, white blood cells, and platelets (PLATE-lets). Red blood cells contain hemoglobin, an iron-rich protein that carries oxygen from your lungs to all parts of your body. Hemoglobin also carries carbon dioxide (a waste gas) from your body to your lungs to be exhaled.
Hemoglobin has two kinds of protein chains: alpha globin and beta globin. If your body doesn't make enough of these protein chains, red blood cells don't form properly and can't carry enough oxygen. Your body won't work well if your red blood cells don't make enough healthy hemoglobin.
Genes control how the body makes hemoglobin protein chains. When these genes are missing or altered, thalassemias occur.
Thalassemias are inherited disorders. That is, they're passed on from parents to their children through genes. People who get abnormal hemoglobin genes from one parent but normal genes from the other are called carriers. Carriers often have no signs of illness other than mild anemia. However, they can pass the abnormal genes on to their children.
People with moderate to severe forms of thalassemia have inherited abnormal genes from both parents.
Alpha Thalassemias
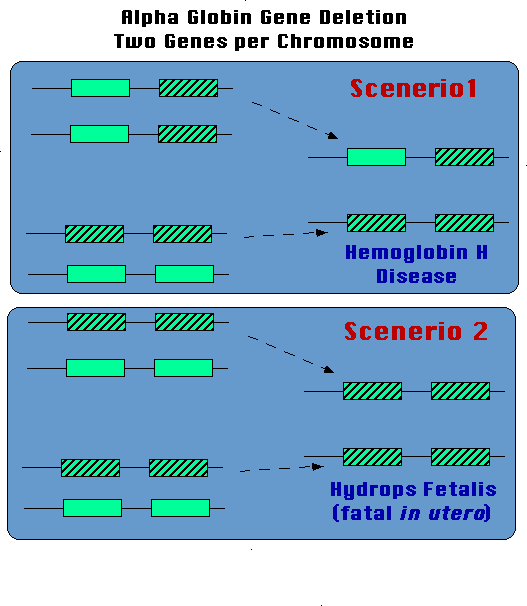
Four genes (two from each parent) are needed to make enough alpha globin protein chains. If one or more of the genes is missing, you will have alpha thalassemia trait or disease. This means that you don't make enough alpha globin protein.
- If you have only one missing gene, you're a silent carrier and won't have any signs of illness.
- If you have two missing genes, you have alpha thalassemia trait (also called alpha thalassemia minor). You may have mild anemia.
- If you have three missing genes, you likely will have hemoglobin H disease (which a blood test can detect). This form of thalassemia causes moderate to severe anemia.
Very rarely, a baby will have all four genes missing. This condition is called alpha thalassemia major or hydrops fetalis. Babies with hydrops fetalis usually die before or shortly after birth.
Inheritance Pattern for Alpha Thalassemia

The diagram shows one example of how alpha thalassemia is inherited. The alpha globin genes are located on chromosome 16. A child inherits four alpha globin genes - two from each parent. In this example, the father is missing two alpha globin genes and the mother is missing one alpha globin gene.
Therefore, each child has a 25 percent chance of inheriting two missing genes and two normal genes (thalassemia trait), three missing genes and one normal gene (hemoglobin H disease), four normal genes (no anemia), or one missing gene and three normal genes (silent carrier).
Beta Thalassemias
Two genes (one from each parent) are needed to make enough beta globin protein chains. If one or both of these genes are altered, you will have beta thalassemia. This means that you don't make enough beta globin protein.
If you have one altered gene, you're a carrier. This condition is called beta thalassemia trait or beta thalassemia minor. It causes mild anemia.
If both genes are altered, you will have beta thalassemia intermedia or beta thalassemia major (also called Cooley's anemia). The intermedia form of the disorder causes moderate anemia. The major form causes severe anemia.
Inheritance Pattern for Beta Thalassemia

The diagram shows one example of how beta thalassemia is inherited. The beta globin gene is located on chromosome 11. A child inherits two beta globin genes—one from each parent. In this example, each parent has one altered beta globin gene.
Therefore, each child has a 25 percent chance of inheriting two normal genes (no anemia), a 50 percent chance of inheriting one altered gene and one normal gene (beta thalassemia trait), or a 25 percent chance of inheriting two altered genes (beta thalassemia major).
Who Is At Risk for Thalassemias?
Family history and ancestry are the two risk factors for thalassemias.
Family History
Thalassemias are inherited, which means they're passed on from parents to their children. If your parents have missing or altered hemoglobin-making genes, you may have a thalassemia.
Ancestry
Alpha thalassemias most often affect people of Southeast Asian, Indian, Chinese, or Filipino origin or ancestry.
Beta thalassemias most often affect people of Mediterranean (Greek, Italian, and Middle Eastern), Asian, or African origin or ancestry.
What Are the Signs and Symptoms of Thalassemias?
Signs and symptoms of thalassemias are due to lack of oxygen in the bloodstream. This occurs because the body doesn't make enough healthy red blood cells and hemoglobin. The severity of symptoms depends on the severity of the disorder.
No Symptoms
Alpha thalassemia silent carriers generally have no signs or symptoms of the disorder. This is because the lack of alpha globin protein is so small that hemoglobin works normally.
Mild Anemia
People who have alpha or beta thalassemia trait can have mild anemia. However, many people with this type of thalassemia have no signs or symptoms.
Mild anemia can make you feel tired. It's often mistaken for iron-deficiency anemia.
Mild to Moderate Anemia and Other Signs and Symptoms
People with beta thalassemia intermedia have mild to moderate anemia. They also may have other health problems, such as:
- Slowed growth and delayed puberty. Anemia can slow down a child's growth and development.
- Bone problems. Thalassemia may make bone marrow (the spongy material inside bones that makes blood cells) expand. This causes wider bones than normal. Bones also may be brittle and break easily.
- An enlarged spleen. The spleen is an organ that helps your body fight infection and remove unwanted material. When a person has a thalassemia, the spleen has to work very hard. As a result, the spleen becomes larger than normal. This makes anemia worse. If the spleen becomes too large, it must be removed.
Severe Anemia and Other Signs and Symptoms
People with hemoglobin H disease or beta thalassemia major (also called Cooley's anemia) have severe thalassemia. Signs and symptoms occur within the first 2 years of life. They may include severe anemia and other serious health problems, such as:
- Pale and listless appearance
- Poor appetite
- Dark urine
- Slowed growth and delayed puberty
- Jaundice (a yellowish color of the skin or whites of the eyes)
- Enlarged spleen, liver, and heart
- Bone problems (especially bones in the face)
Better treatments now allow people who have moderate and severe thalassemias to live much longer. As a result, these people must cope with complications of the disorder that occur over time.
Heart and Liver Disease
Regular blood transfusions are a standard treatment for thalassemias. As a result, iron can build up in the blood. This can damage organs and tissues, especially the heart and liver.
Heart disease caused by iron overload is the main cause of death in people who have thalassemias. Heart disease includes heart failure, arrhythmias (irregular heartbeats), and heart attack.
Infection
Among people who have thalassemias, infections are a key cause of illness and the second most common cause of death. People who have had their spleens removed are at even higher risk, because they no longer have this infection-fighting organ.
Osteoporosis
Many people who have thalassemias have bone problems, including osteoporosis (OS-te-o-po-RO-sis). This is a condition in which bones are weak and brittle and break easily.
How Are Thalassemias Diagnosed?
Doctors diagnose thalassemias using blood tests, including a complete blood count (CBC) and special hemoglobin tests.
- A CBC provides information about the amount of hemoglobin and the different kinds of blood cells, such as red blood cells, in a sample of blood. People who have thalassemias have fewer healthy red blood cells and less hemoglobin in their blood than normal. People who have alpha or beta thalassemia trait may have smaller than normal red blood cells.
- Hemoglobin tests measure the types of hemoglobin in a blood sample. People who have thalassemias have problems with the alpha or beta globin protein chains of hemoglobin.
Moderate and severe thalassemias usually are diagnosed in early childhood. This is because signs and symptoms, including severe anemia, appear within the first 2 years of life.
People who have milder forms of thalassemia may be diagnosed after a routine blood test shows they have anemia. Doctors suspect thalassemia if a child has anemia and is a member of an ethnic group that's at increased risk for thalassemia.
Doctors also do tests on the amount of iron in the blood to find out whether the anemia is due to iron deficiency or thalassemia. Iron-deficiency anemia occurs when the body doesn't have enough iron to make hemoglobin. The anemia in thalassemia occurs because of a problem with either the alpha globin chain or the beta globin chain of hemoglobin, not because of a lack of iron.
Because thalassemias are passed on from parents to children, family genetic studies also can help diagnose the disorder. This involves taking a family medical history and doing blood tests on family members to show whether any have missing or altered hemoglobin genes.
If you know of family members who have thalassemias and you're thinking of having children, consider talking with your doctor and/or a genetic counselor. They can help determine your risk for passing on the disorder to a child.
If you're expecting a baby and you and your partner are thalassemia carriers, you may want to consider prenatal testing.
Prenatal testing involves taking a sample of amniotic fluid or tissue from the placenta. (Amniotic fluid is the fluid in the sac surrounding a growing embryo. The placenta is the organ that attaches the umbilical cord to the mother's womb.) Tests done on the fluid or tissue can show whether your baby has thalassemia and how severe it's likely to be.
How Are Thalassemias Treated?
Treatments for thalassemias depend on the type and severity of the disorder. People who are carriers or who have alpha or beta thalassemia trait have mild or no symptoms. They need little or no treatment.
Doctors use three standard treatments for moderate and severe forms of thalassemia. These include blood transfusions, iron chelation (ke-LAY-shun) therapy, and folic acid supplements. Other treatments have been developed or are being tested, but they're used much less often.
Standard Treatments
Blood Transfusions
Transfusions of red blood cells are the main treatment for people who have moderate or severe thalassemias. A blood transfusion, given through a needle in a vein, gives you healthy red blood cells with normal hemoglobin. Red blood cells live for only about 120 days. So, you may need repeated transfusions to maintain a supply of healthy red blood cells.
If you have hemoglobin H disease or beta thalassemia intermedia, you may need blood transfusions on occasion. For example, you may need this treatment when you have an infection or other illness, or when your anemia is severe enough to cause tiredness.
If you have beta thalassemia major, or Cooley's anemia, you need regular blood transfusions (often every 2 to 4 weeks). These will help you maintain normal hemoglobin levels and red blood cell numbers. Blood transfusions allow you to feel better, enjoy normal activities, and live into adulthood.
Blood transfusions are lifesaving, but they're expensive and carry a risk of transmitting infections and viruses (for example, hepatitis). However, this risk is very low in the United States because of careful blood screening.
Iron Chelation Therapy
Because the hemoglobin in red blood cells is an iron-rich protein, regular blood transfusions can lead to a buildup of iron in the blood. This condition is called iron overload. It damages the liver, heart, and other parts of the body.
To prevent this damage, iron chelation therapy is needed to remove excess iron from the body. Two medicines are used for iron chelation therapy.
- Deferoxamine (Desferal) is a liquid medicine that's given slowly under the skin, usually with a small portable pump used overnight. This therapy takes time and can be mildly painful. Side effects include loss of vision and hearing.
- Deferasirox is a pill taken once a day. Side effects include headache, nausea (feeling sick to the stomach), vomiting, diarrhea, joint pain, and fatigue (tiredness).
Folic Acid Supplements
Folic acid is a B vitamin that helps build healthy red blood cells. You may need to take folic acid supplements in addition to blood transfusions and/or iron chelation therapy.
Other Treatments
Other treatments have been developed or are being tested, but they're used much less often.
Blood and Marrow Stem Cell Transplant
A blood and marrow stem cell transplant replaces your abnormal or faulty stem cells with healthy ones from another person (a donor). Stem cells are the cells inside bone marrow that make red blood cells and other types of blood cells.
A stem cell transplant is the only treatment that can cure thalassemia. But only a small number of people who have the severe form of the disorder are able to find a good match among donors and have the risky procedure.
Possible Future Treatments
Researchers are working to find new treatments for thalassemias. For example, it may be possible someday to insert a normal hemoglobin gene into stem cells in bone marrow. This will allow people to make their own healthy red blood cells and hemoglobin.
Researchers also are studying ways to trigger a person's ability to make fetal hemoglobin after birth. This type of hemoglobin is found in fetuses and newborns. After birth, the body switches to making adult hemoglobin. Making more fetal hemoglobin may make up for the lack of healthy adult hemoglobin.
Treating Complications
Better treatments now allow people who have moderate and severe thalassemias to live much longer. As a result, these people must cope with complications that occur over time.
An important part of managing thalassemias is treating complications. Treatment may be needed for heart or liver diseases, infections, osteoporosis, and other problems.
Can Thalassemias Be Prevented?
Thalassemias can't be prevented because they're inherited (passed on from parents to children). However, these bleeding disorders can be found before birth through prenatal tests.
Family genetic studies may help find out whether people have missing or altered hemoglobin genes that cause thalassemias.
If you know of family members who have thalassemias and you're thinking of having children, consider talking with your doctor and/or a genetic counselor. They can help determine your risk for passing on the disorder to your child.
Living With Thalassemias
Survival and quality of life are now much better for people who have moderate or severe forms of thalassemia. This is because:
- More people are able to get blood transfusions now.
- Blood screening has reduced the number of infections from blood transfusions. Also, treatments for other kinds of infections have improved.
- New iron chelation treatments are available that are easier for some people to take.
- Some people have been cured through blood and marrow stem cell transplants.
Living with thalassemia can be challenging, but several approaches can help you cope.
Follow Your Treatment Plan
It's important to follow the treatment plan your doctor gives you. Get blood transfusions as he or she recommends.
Take your iron chelation medicine. This is important because the leading cause of death among people with thalassemias is heart disease caused by iron overload. Iron buildup can damage your heart, liver, and other organs. Although the iron chelation treatment can take time and be mildly painful, it's important that you don't stop taking your medicine.
Several chelation treatments are now available, including injections and pills. Your doctor will talk to you about which treatment is best for you.
Take folic acid supplements if your doctor prescribes them. Folic acid is a B vitamin that helps build healthy red blood cells.
Get Ongoing Medical Care
It's important that you keep your scheduled medical appointments and get any tests that your doctor recommends.
These tests may include:
- Monthly complete blood counts, and tests for blood iron levels every 3 months
- Yearly tests for heart function, liver function, and viral infection (for example, hepatitis B and hepatitis C and HIV)
- Yearly tests to check for iron buildup in your liver
- Yearly vision and hearing tests
- Regular checkups to make sure blood transfusions are working
- Other tests as needed (such as lung function tests, genetic tests, and tests to match your tissues against a possible donor if a stem cell transplant is being considered)
Children who have thalassemias should receive yearly checkups to monitor their growth and development. The checkup includes a physical exam, including a height and weight check, and any necessary tests.
Take Measures To Stay Healthy
Take steps to stay as healthy as possible. Follow a healthy eating plan. Follow your doctor's instructions about taking iron supplements.
Get vaccinations as needed, especially if you've had your spleen removed. You may need vaccinations for flu, pneumonia, hepatitis B, and meningitis. Your doctor can tell you which vaccines you need.
Watch for signs of infections (such as fever) and take steps to lower your chance of getting an infection. This is especially important if you've had your spleen removed.
- Wash your hands often.
- Avoid crowds during cold and flu season.
- Keep the skin around the site where you get blood transfusions as clean as possible.
- Call your doctor if fever develops.
Seek Help and Support
Joining a support group may help you cope with thalassemia if you or your child has it. Talking to others who live with the same issues can help you see how they've coped with them. To find a local support group, contact the Cooley's Anemia Foundation.
Thalassemias At A Glance
- Thalassemias are inherited blood disorders. "Inherited" means they're passed on from parents to children.
- Thalassemias cause the body to make fewer healthy red blood cells and less hemoglobin than normal.
- People who have thalassemias can have mild or severe anemia. This condition is caused by a lower than normal number of red blood cells or not enough hemoglobin in the red blood cells.
- The two major types of thalassemia are alpha thalassemia and beta thalassemia. There are different forms of each type.
- Thalassemias occur when the genes that control the production of hemoglobin are missing or altered. Your body won't work properly if your red blood cells don't make enough healthy hemoglobin.
- Family history and ancestry are the two risk factors for thalassemias. If your parents have missing or altered hemoglobin-making genes, you may have thalassemia. Thalassemias occur most often among people of Italian, Greek, Middle Eastern, Asian, and African descent.
- Signs and symptoms of thalassemias are due to lack of oxygen in the bloodstream. They may include mild to severe anemia; slowed growth and delayed puberty; bone problems; and enlarged spleen, liver, or heart; a pale and listless appearance; poor appetite; dark urine; and jaundice (a yellowish color of the skin or whites of the eyes). Symptoms depend on the type of thalassemia you have. Silent carriers have no symptoms.
- Doctors diagnose thalassemias using blood tests, including a complete blood count and special hemoglobin tests. Prenatal testing can show whether an unborn baby has thalassemia and how severe it's likely to be.
- People who have mild thalassemia often need little or no treatment. There are three standard treatments for moderate and severe forms of thalassemia. These include blood transfusions, iron chelation therapy, and folic acid supplements.
- Better treatments now allow people who have moderate and severe thalassemias to live much longer. As a result, these people must cope with complications of the disease that develop over time. Complications include heart and liver disease, infections, osteoporosis, and other problems.
- Thalassemias can't be prevented because they're inherited. However, these blood disorders can be found before birth through prenatal testing.
- Living with thalassemia can be challenging, but several approaches can help you cope. Follow your doctor's treatment plan, get ongoing medical care, take measures to stay healthy, and seek help and support.
Other names for thalassemia
The various types of thalassemia have specific names related to the severity of the disorder.
Alpha Thalassemias
- Alpha thalassemia silent carrier
- Alpha thalassemia minor, also called alpha thalassemia trait
- Hemoglobin H disease
- Alpha thalassemia major, also called hydrops fetalis
Beta Thalassemias
- Beta thalassemia minor, also called beta thalassemia trait
- Beta thalassemia intermedia
- Beta thalassemia major, also called Cooley's anemia or beta-zero (β0) thalassemia
- Beta-plus (β) thalassemia
- Mediterranean anemia
SOURCE: National Heart Lung and Blood Institute
|
Bookmark this post:
|
|
2 comments
Here is a link to more information about the genetics of Beta Thalassemia that was prepared by our genetic counselor and which has links to some useful resource for those dealing with this condition: http://www.accessdna.com/condition/Beta_Thalassemia/60. There is also a number listed for anyone who wants to speak to a genetic counselor by phone. I hope it helps. Thanks, AccessDNA
Posted on February 6, 2010 at 1:55 PM
thanks for the comment
Posted on February 7, 2010 at 5:22 AM
Post a Comment